X-ray Crystallography: A Gold Standard for Precision in Structural Biology
X-ray crystallography remains one of the most powerful and widely used techniques for determining the three-dimensional (3D) structures of biomolecules, particularly proteins. As a cornerstone method in structural biology, it has enabled the detailed exploration of molecular architectures, providing insights into how biological macromolecules function at the atomic level. Below, we will explore why X-ray crystallography is often considered the "gold standard" for precision in structural determination and its ongoing importance in research and medicine.
✽ How X-ray Crystallography Works
X-ray crystallography involves the following key steps:
Protein Crystallization: The first challenge is obtaining high-quality single crystals of the protein or macromolecule of interest. The quality and size of the crystal are crucial for successful data collection. Crystals must be large enough and regular in structure so that the X-ray beams interact with them in a way that produces diffraction patterns.
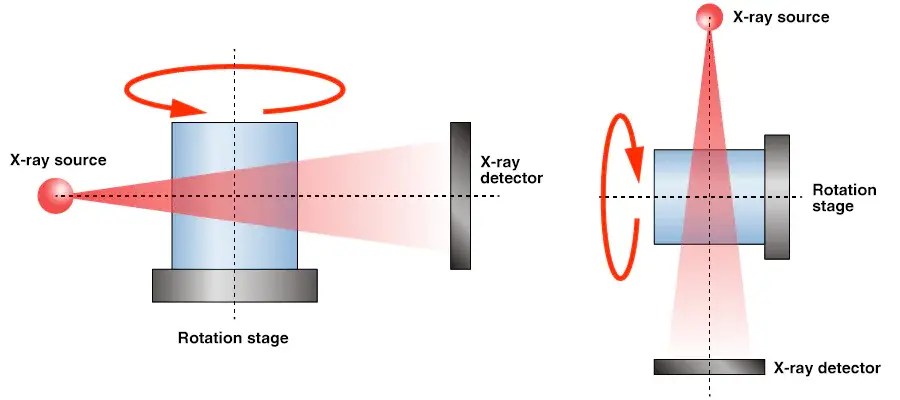
X-ray Diffraction: Once a suitable crystal is obtained, it is exposed to X-rays. The X-rays interact with the electrons in the crystal, causing them to diffract in various directions. The diffraction pattern produced contains information about the atomic arrangement within the crystal.
Data Collection and Analysis: The diffraction pattern is captured by a detector, and the resulting data are used to calculate the electron density map. This map represents the probability distribution of electrons within the molecule, providing the framework for determining the positions of atoms in the 3D structure.
Model Building and Refinement: Using the electron density map, scientists build an atomic model of the protein or molecule. The model is then refined by adjusting it to fit the experimental data more accurately. This iterative process results in a highly precise structure that reveals the spatial arrangement of atoms, which is crucial for understanding the molecule’s function.
Structure Validation: The final structure is carefully validated by comparing it against known experimental and theoretical parameters, ensuring its accuracy. This can include checking for stereochemistry, bond angles, and other physical characteristics.
✽ Why X-ray Crystallography is the "Gold Standard"
- Atomic Resolution: X-ray crystallography can provide extremely high-resolution structures, often down to 1.0–1.5 Å (angstroms). This allows researchers to observe the positions of individual atoms in the protein, providing unprecedented insight into its conformation and function. This level of precision is critical for understanding the detailed molecular interactions that underpin biological processes.
- Reproducibility and Accuracy: When carried out correctly, X-ray crystallography offers highly reproducible and accurate results. Given its ability to directly probe the electron density within crystals, it provides a definitive representation of the molecule’s 3D structure, often seen as more reliable than other techniques like NMR spectroscopy or cryo-EM for small to medium-sized proteins.
- Wide Range of Applications: X-ray crystallography is versatile and can be applied to a broad range of biological molecules, including proteins, nucleic acids, and small molecules. It is particularly useful for studying enzymes, receptor-ligand interactions, and complex macromolecular assemblies, which are often too large or too flexible for other structural techniques.
- Pharmaceutical Applications: In drug design, X-ray crystallography has been instrumental in the development of targeted therapies. By providing detailed structural information about the active sites of enzymes and receptors, researchers can design molecules that specifically interact with these sites, leading to more effective and selective drugs. Examples include the development of HIV protease inhibitors and various cancer therapies targeting protein kinases.
- Unparalleled Precision in Atomic Details: The detailed atomic information provided by X-ray crystallography is unmatched by other techniques, such as NMR or cryo-EM. This allows for a deeper understanding of how proteins fold, how they interact with ligands, and how mutations may affect their function. In structural genomics, where large-scale determination of protein structures is pursued, X-ray crystallography has been crucial for uncovering the structures of many "unknown" proteins.
Limitations and Challenges
While X-ray crystallography remains a gold standard, it is not without its challenges:
- Crystallization Difficulty: One of the biggest obstacles in X-ray crystallography is obtaining high-quality, large single crystals. Many proteins, especially membrane proteins, are difficult to crystallize, and some cannot form stable crystals at all. This limitation has prompted the development of complementary techniques, like cryo-electron microscopy (cryo-EM), which does not require crystallization.
- Static Structures: X-ray crystallography captures a "snapshot" of the protein in a single conformation, which may not fully represent its behavior in a dynamic cellular environment. Proteins often undergo conformational changes in response to binding ligands or other cellular signals, and these movements may not be captured in static crystal structures.
- Crystallization Bias: The crystallization process can sometimes select for certain conformations of the protein over others, which means the structure obtained might not represent the protein’s most biologically relevant state. This is particularly problematic for proteins with flexible regions or for those that exist in multiple conformations.
Despite these challenges, X-ray crystallography is often used in conjunction with other structural techniques to overcome its limitations:
- Cryo-Electron Microscopy (cryo-EM): Cryo-EM has emerged as a powerful alternative for studying large, dynamic complexes, and for molecules that are difficult to crystallize. Cryo-EM can provide structural information at near-atomic resolution without the need for crystals, making it a complementary technique to X-ray crystallography.
- Nuclear Magnetic Resonance (NMR): NMR is ideal for studying smaller proteins or protein fragments in solution. While it offers atomic-level information similar to X-ray crystallography, it typically provides lower resolution and is limited to smaller systems.
- Small-Angle X-ray Scattering (SAXS): SAXS provides information about the overall shape and size of proteins in solution, helping to capture conformational changes that may not be detected by X-ray crystallography alone.

Impact on Drug Discovery and Development
The precision offered by X-ray crystallography has revolutionized drug discovery. By determining the 3D structures of target proteins, researchers can:
- Identify Binding Sites: Structural data helps identify the exact location and shape of binding sites on proteins, allowing for the rational design of molecules that can precisely target these sites.
- Design Inhibitors: X-ray crystallography is particularly useful in the design of enzyme inhibitors or receptor antagonists, as it allows for the design of small molecules that fit perfectly into the active site or binding pocket.
- Screen for Drug Candidates: High-throughput screening can be combined with crystallographic data to identify drug-like molecules that bind to the target with high affinity, streamlining the drug discovery process.
X-ray crystallography continues to be a gold standard in structural biology, offering unmatched precision and accuracy in determining the 3D structures of proteins and other biomolecules. Despite its challenges, it remains an essential tool in understanding molecular mechanisms and in the development of new therapeutic strategies. As technology advances, particularly with the integration of cryo-EM and computational modeling, X-ray crystallography will continue to play a critical role in uncovering the molecular basis of health and disease, driving forward innovations in medicine.